Recurrent Acute Stent Thrombosis and Myocardial Infarction in a Young Patient
John Evans, D.O., Stephen Dixon, D.O., and Sharolyn Cook, D.O.
Abstract
Myocardial infarction in patients under the age of 45 is considered rare, occurring in approximately 6-10% of the general population.1,2 In many cases, premature coronary atherosclerosis plays an important causative role, however about 20% of young adult coronary thrombosis can be attributed to hypercoagulability.3 In this article, we present a 43-year-old male with recurrent coronary thrombosis. While an initial presentation of acute myocardial infarction appeared driven by common atherogenic risk factors of tobacco use and diabetes, subsequent presentation with coronary re-thrombosis and acute stent thrombosis revealed a hypercoagulable state. This case highlights challenges in management of acute myocardial infarction and hypercoagulable states. It emphasizes the vigilance that must be exercised in generating differential diagnoses for coronary thrombosis.
Introduction
Inherited hypercoagulability appears infrequently in general medical practice and is thought to have a prevalence of less than 1 in 1000, although if present, it may lead to life threatening organ failure including myocardial infarction, renal failure, gastrointestinal, and respiratory symptoms.4,5 Diagnosis of a primary hypercoagulable state is suggested by recurrent thrombosis and laboratory abnormalities that include the presence of either a procoagulant molecule or derangement of the clotting mechanism. Well-described secondary causes of hypercoagulability can include medications such as procainamide (which may induce a lupus-like syndrome), autoimmune disease, and infection. Treatment includes antithrombotic medications such as a vitamin K antagonist, immunosuppressants, or plasmapheresis.6 Even when traditional atherosclerotic mechanisms are known in a patient with myocardial infarction, hypercoagulability may be a dominant, even synergistic, contributor to thrombotic risk. 7,8 We present a case of recurrent coronary thrombosis in the setting of probable catastrophic antiphospholipid antibody syndrome that highlights the therapeutic challenges inherent in mitigating both short and long term complications in a hypercoagulable state.
Case Report
A 43-year-old male presented with non-ST segment elevation acute coronary syndrome and underwent urgent coronary angiography and attempted percutaneous revascularization. The past medical history included an acute anterior wall myocardial infarction (MI) approximately two years earlier with ischemic cardiomyopathy requiring implantable cardioverter defibrillator insertion. Medical history was notable for chronic kidney disease stage 4, diabetes mellitus with insulin use, hypertension, dyslipidemia, and a prominent history of tobacco use for which he had remained abstinent in the past 48 months. The presenting symptoms included substernal, dull, non-radiating chest pain and dyspnea. Peak troponin I elevation was 20.0 ng/dL (normal range 0-0.04 ng/dL) and a resting electrocardiogram did not show ST segment elevations (Figure 1).
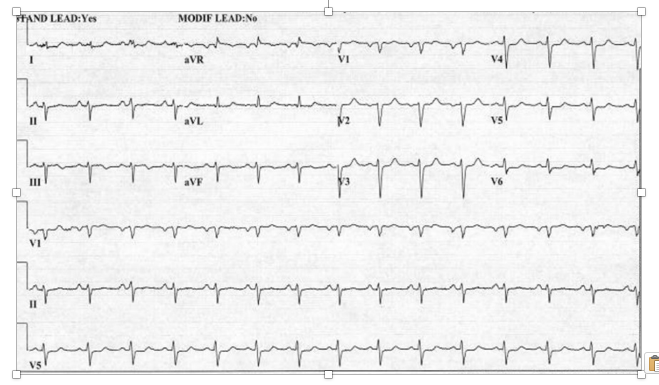
Figure 1. Resting electrocardiogram of a 43-year-old male presenting with acute non-ST segment elevation myocardial infarction caused by acute right coronary thrombus
Serum creatinine at admission was elevated to 4.25 mg/dL (normal range 0.70-1.30). Antithrombotic therapies were begun at hospital admission including weight-based heparin and later initiation of a glycoprotein IIbIIIa (GPIIbIIIa) inhibitor, eptifibatide. The patient had remained compliant with ticagrelor and aspirin following the previous MI, and these were continued in the hospital. Urgent coronary angiography was performed due to ongoing ischemic symptoms despite medical therapy. Coronary angiography revealed thrombotic occlusion of the proximal right coronary artery (RCA) (Figure 2a).
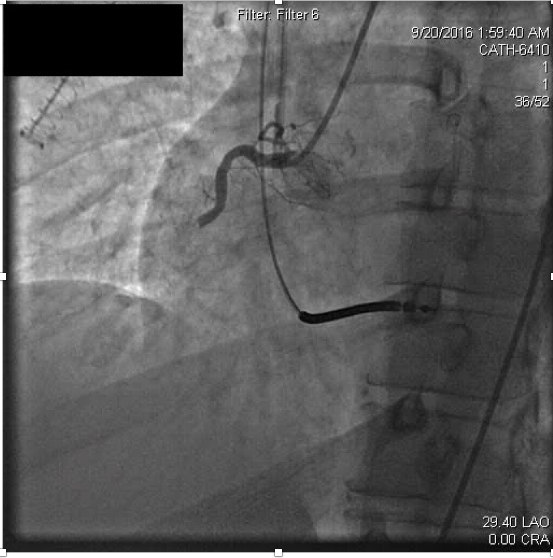
Figure 2a. Acute thrombosis within the proximal right coronary artery.
Anticoagulation was continued with weight based boluses of heparin to achieve an activated clotting time (ACT) of ≥250. Percutaneous intervention was attempted with suction thrombectomy, balloon angioplasty, and drug-eluting stent delivery through the mid to distal right coronary artery. Three everolimus-eluting stents were deployed in overlapping fashion and dilated to nominal atmospheres. Stent apposition was then interrogated with intravascular ultrasound (IVUS), and each stent was determined to be appropriately deployed after post-dilation to high atmospheres (16-18 atm). Following this, coronary occlusion persisted, but he was free of chest discomfort and remained hemodynamically and electrically stable (Figure 2b).
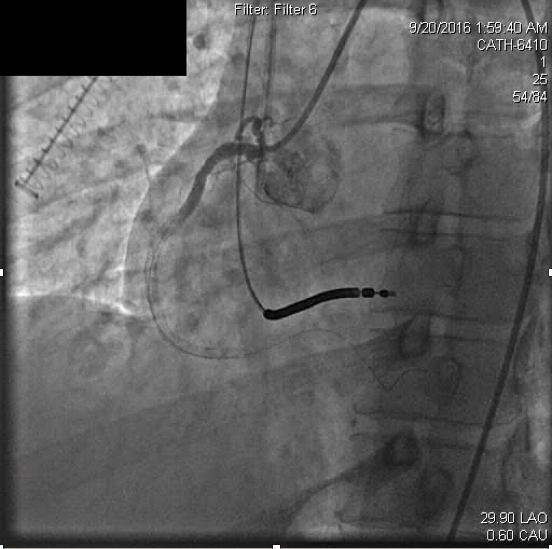
Figure 2b. Persistent coronary thrombosis following attempted percutaneous coronary intervention
Due to concerns for contrast exposure with acute renal failure and lack of perceived benefit to the patient, attempted revascularization was abandoned and he was transferred to the intensive care unit for close monitoring. Dual antiplatelet therapy was continued as before with aspirin 81mg and ticagrelor 90mg twice daily. In the following 24 hours, however, he experienced recurrent angina and was taken back to the catheterization laboratory. Remarkably, the previously occluded RCA appeared recanalized but with thrombus apparent at the distal stent edge at the ostium of the right posterolateral branch (Figure 3a).
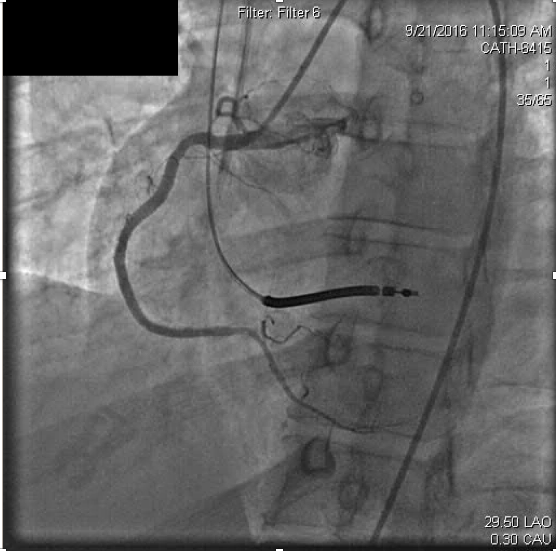
Figure 3a. On repeat right coronary angiography for acute , acute thrombosis is seen within the distal right coronary stent.
Slow injection of intracoronary abciximab (14mg over multiple injections) was administered with resolution of the thrombus and ischemic symptoms (Figure 3b).
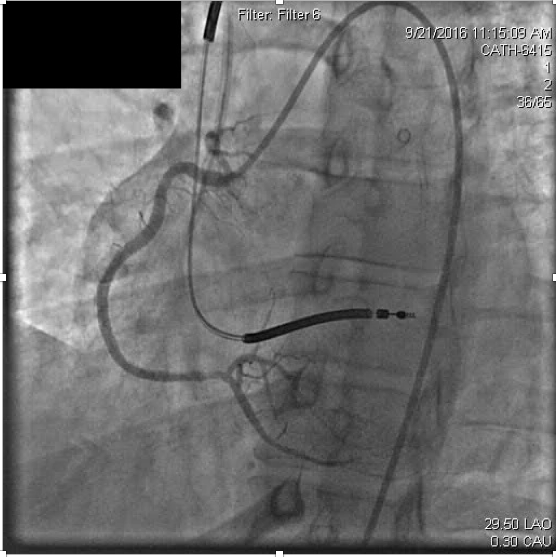
Figure 3b. After recanalization of the distal right coronary stent, the occluded right posterolateral branch is visualized.
The patient was then loaded orally with a reduced dose of prasugrel 30mg, and IV abciximab was continued post-procedure after returning to the intensive care unit. A reduced loading dose of prasugrel 30mg was chosen due to concerns for bleeding, having previously received ticagrelor, heparin, and abciximab. Left ventricular (LV) function assessment by echocardiogram later revealed severely reduced LV function with anteroseptal and apical akinesis with an estimated ejection fraction of 25%.
Laboratory workup including a hypercoagulable panel revealed marked elevation in beta-2 microglobulin I IgG at 71.4 specific gravity units (SGU) (normal range 0-19.9) and anticardiolipin IgG antibodies at 101.6 U/mL (normal range 0-15). Anti-neutrophil cytoplasmic antibodies (ANCA) were negative and the antinuclear antibody screen was positive (1:320 with a homogeneous pattern). An extractable nuclear antigen panel was normal as were hepatitis serologies. No mutations were found in either Factor II or V. Based on these laboratories and clinical presentation, a presumptive diagnosis of antiphospholipid antibody syndrome (APL) was made. The anti-Xa medication apixaban 5mg twice daily was initiated after consultation with the hematology service. Renal function did not normalize with oliguria noted. Renal biopsy was discussed but deferred due to the necessity of uninterrupted antiplatelet therapy with recent coronary stents. Consultation with the nephrology service was obtained and hemodialysis was initiated routinely. The patient was then discharged home but returned within 3 months for a complaint of fatigue, malaise, and lower extremity palpable purpuric rash ascending from both feet proximally. In the interim between hospital admissions, renal function had improved such that he was able to discontinue hemodialysis, but he remained with severely reduced renal function. Beta-2-microglobulin I IgG serology was repeated with marked persistent elevation (72.4 SGU). Again, renal biopsy was considered but not performed due to the necessity of antiplatelet therapies and risk of bleeding. Skin biopsy was performed and revealed a leukocytoclastic vasculitis that was IgA negative by immunofluorescence (Figure 4).
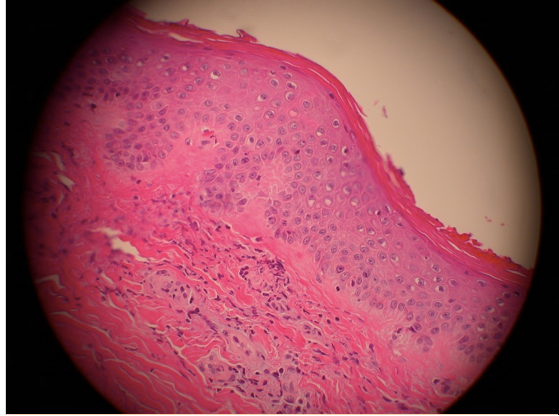
Figure 4. Skin biopsy specimen reveals vasculitis with occluded capillaries (arrow) and leukocytic infiltrate.
Laboratory and biopsy findings supported a diagnosis of antiphospholipid antibody syndrome. He complained of chest pain which was atypical in comparison with previous anginal complaints and underwent regadenoson myocardial perfusion stress imaging. Imaging noted prior inferior and apical infarcts but no areas of reversible ischemia. A regimen of high-dose IV methylprednisolone was initiated and (1,000mg times 3 doses) the purpura resolved, but renal function did not return to normal. He was again discharged home with outpatient hemodialysis, continued antithrombotic and antiplatelet therapies, and an oral prednisone regimen of 10mg daily.
Discussion
Hypercoagulable states are recognized causes of venous and arterial thrombosis.9,10 In a large case control study of approximately 8,000 male patients with coronary disease and stroke, the presence of elevated beta-2-glycoprotein 1-dependent anticardiolipin antibodies was associated with increased risk of arterial thrombosis.11 Additional studies have highlighted a synergistic role for atherogenesis and hypercoagulability in myocardial infarction.12 As with other patients who present with acute myocardial infarction, therapy should be primarily directed at revascularization, with both primary angioplasty and thrombolysis playing a role.13,14 The role of warfarin is well described, supported in the literature, and remains a cornerstone therapy with a goal INR of 2.0-3.0.15,16 A study using the novel oral anticoagulant rivaroxaban in antiphospholipid antibody syndrome is ongoing with early results revealing a trend of non-inferiority compared to warfarin.17 Acute re-thrombosis, particularly in-stent, remains a risk after percutaneous intervention in patients with APL and should be managed with prompt initiation of antithrombotic medication in addition to appropriate dual antiplatelet therapy.18 Our case details the therapeutic challenges of managing thrombotic risk in the setting of APL and acute myocardial infarction. Maintaining a high degree of suspicion for underlying hypercoagulable states is paramount in those patients with premature acute or recurrent thrombotic syndromes.
References
1. Hong MK, Cho SY, Hong BK, et al. Acute myocardial infarction in the young adults. Yonsei Medical Journal. 1994;35:184–189
2. Jalowiec DA, Hill, JA. Myocardial infarction in the young and in women. Cardiovascular Clinics. 1989;20:197–206.
3. Choudhury L, Marsh JD. Myocardial Infarction in Young Patients. American Journal of Medicine. 1999;107(3):254-261.
4. Lane DA, Mannuci PM, Bauer KA, et al. Inherited Thrombophilia: Part 1. Article in Thrombosis and Haemostasis. 1996;76(5):651-662.
5. Nachman RL, Silverstein R. Hypercoagulable States. Annals of Internal Medicine. 1993;119:819-827.
6. Bick RL, Kaplan H. Syndromes of Thrombosis and Hypercoagulability: Congenital and Acquired Causes of Thrombosis. Medical Clinics of North America. 1998;82(3):409-458.
7. Inbal, A., Freimark, D., Modan, B., Chetrit, A., Matetzky, S., Rosenberg, N., Dardik, R., Baron, Z., & Seligsohn, U. Synergistic Effects of Prothrombotic Polymorphisms and Atherogenic Factors on the Risk of Myocardial Infarction in Young Males. Blood. 1999;93(7):2186-2190.
8. Batalla A, Reguero JR, Cubero GI, Coto E. Prothrombotic Genetic Risk Factors and Myocardial Infarction at Young Age. Blood. 1999;94:3959-3960.
9. Kannel WB, Abbott RD (1984) Incidence and prognosis of unrecognized myocardial infarction. An update on the Framingham study. N Engl J Med 311:1144–1147.
10. Levy PJ, Gonzalez FM, Rush DS, Haynes JL. Hypercoagulable states as an evolving risk for spontaneous venous and arterial thrombosis. J Am Coll Surg. 1994 Mar;178(3):266-70
11. Brey RL, Abbott RD, Sharp DS, Ross GW, Curb JD, Stallworth CL, Kittner SJ. Beta-2-glycoprotein 1-dependent (B2GP1-dep) Anticardiolipin Antibodies (aCL) Are An Independent Risk Factor for Ischemic Stroke in the Honolulu Heart Cohort. Stroke. 2001;32:1701-1706.
12. Doggen CJM, Cats VM, Bertina RM, Rosendaal FR. Interaction of coagulation defects and cardiovascular risk factors. Increased risk of myocardial infarction associated with factor V Leiden or prothrombin. Circulation. 1998;97:1037.
13. Ho YL, Chen MF, Wu CC, et al. Successful treatment of acute myocardial infarction by thrombolytic therapy in a patient with primary antiphospholipid antibody syndrome. Cardiology.1996;87:354–357.
14. Takeuchi S, Obayashi T, Toyama J. Primary antiphospholipid syndrome with acute myocardial infarction recanalised by PTCA. Heart. 1998;79:96–98.
15. Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. The Lancet. 2010;376(9751):1498-1509.
16. Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Huges GRV. The management of Thrombosis in the Antiphospholipid-Antibody Syndrome. The New England Journal of Medicine. 1995;332(15):993-997.
17. Cohen H, Hunt BJ, Efthymiou M, Arachchillage DRJ, Mackie IJ, et al. Rivaroxaban versus warfarin to treat patients with thrombotic antiphospholipid syndrome, with or without systemic lupus erythematosus (RAPS): a randomised, controlled, open-label, phase 2/3, non-inferiority trial. Lancet Haematology.2016;3:426-436.
18. Koniari I, Siminelakis SN, Baikoussis NG, et al. Antiphospholipid syndrome; its implication in cardiovascular diseases: a review. Journal of Cardiothoracic Surgery. 2010;5(101).