Ewing Sarcoma; Suprasellar Mass in a Pediatric Patient
Aaron Bush D.O., Oklahoma State University Medical Center
Yoon Cho D.O., Oklahoma State University Medical Center
Weston Zickgraf D.O., Oklahoma State University Medical Center
Donald von Borstel D.O. Oklahoma State University Medical Center
Funding associated with this manuscript: No funding was received to produce this manuscript.
Conflicts of Interest: No conflicts of interest to disclose with the production of this manuscript.
Key Words: Ewing Sarcoma, Suprasellar Mass, Pediatrics
Abstract
A common differential diagnosis within the field of neuroradiology is the potential
etiologies of a suprasellar mass. Unlike in the adult population, there is a limited differential
diagnosis when considering suprasellar masses in children1. This differential includes
craniopharyngiomas, germinomas, hypothalamic low-grade glioma, hypothalamic hamartoma,
and Langerhans cell histiocytosis. While many of these lesions are benign in nature, some have
malignant potential and determining the proper work-up and evaluation of these lesions is
critical. In this article, we discuss a pediatric patient who presents with symptoms of mass effect
due to a suprasellar mass causing vision loss and headaches. The patient was diagnosed via CT
and MRI in addition to endoscopic evaluation and biopsy by an ENT physician. This case report
focuses on the imaging and differential considerations in these patients, as well as additional
points to consider in the evaluation and management of patients with Ewing Sarcoma.
Introduction
Ewing sarcoma is a group of highly malignant tumors involving a chromosomal
abnormality that results in abnormal growth of osseous structures2. While these often present
within the diaphysis of long bones in patients aged 4-25-years-old, they can occur in any
location and age population. Ewing sarcoma is the second most common malignant tumor,
following osteosarcoma, derived from undifferentiated mesenchymal cells of the bone marrow
or neuroectodermal cells (small round blue cell tumor)3. Presenting symptoms are often pain, fever, leukocytosis, and elevated ESR mimicking an infectious etiology. Prognosis is poor with a 5 year survival rate of approximately 40%.
Case Report
Our patient is an 11-year-old female without significant past medical history who
presented with 4-weeks of progressive vision loss and worsening headache. The patient denied
a recent history of trauma or injury to the region. On physical exam, the patient had numbness
to palpation around the left eye. At first, the patient was started on steroid treatments with
Decadron, but the patient's symptoms failed to improve. A CT of the brain was performed
revealing a suprasellar mass involving the left orbit (Figure 1). Subsequently, a follow up MRI of
the brain and orbits with and without intravenous contrast was performed and demonstrated a
sphenoid mass extending to the posterior left orbit (Figure 2). Direct visualization by ENT was
performed via endoscopic evaluation (Figure 3), which showed a large mass protruding from the
posterior ethmoidal air cell into the anterior ethmoidal air cell with complete erosion of the
posterior ethmoid air cells. A punch biopsy was performed and pathology showed a small round
blue cell tumor consistent with an Ewing sarcoma (Figure 4).
Follow-up PET/CT was performed that showed the left sphenoid mass with extension
into the medial wall of the right orbit, suggesting the mass was extending across the midline
(Figure 5). Bilateral bone marrow biopsies, port placement, and echocardiogram were then
performed for staging and chemotherapy planning. Ophthalmology was also consulted and
determined the patient had a new afferent pupillary defect in addition to ptosis and motility
impingement that had worsened from the week prior. Due to rapidly worsening symptoms,
emergent chemotherapy was initiated with doxorubicin, cyclophosphamide, and vincristine.
Repeat MRI showed continued progression of the tumor into the left orbit and encasing
the left cavernous internal carotid artery. Final pathology results were consistent with Ewing’s
sarcoma. Patient was discharged with chemotherapy and radiation therapy planning.
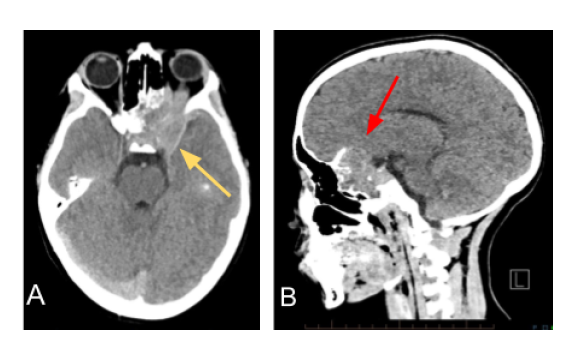
Figure 1: Non-contrast enhanced CT axial (A.) and sagittal (B.) planes demonstrating a mass
(yellow arrow) centered in the left sphenoid extending into the superior and posterior left orbit.
Additionally, there is cortical destruction (red arrow) and no evidence of internal mineralization.
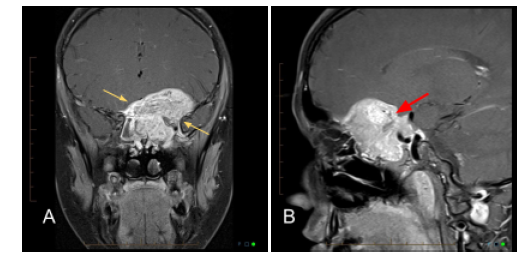
Figure 2: Coronal (A) and sagittal (B) post-contrast enhanced MRI T1 fat-saturated sequences
demonstrating a large mass centered in the left sphenoid (yellow arrows) extending into the
superior and posterior left orbit with multiple internal flow voids (red arrow). The mass
enhances avidly on post-contrast imaging.
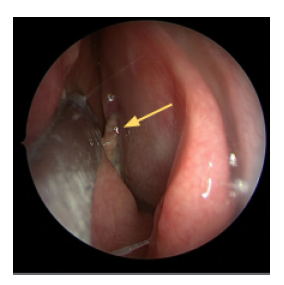
Figure 3: Intraoperative endoscopic image of the left nasal cavity. A tan/white mass (yellow
arrow) was visualized protruding from the posterior ethmoidal air cell.
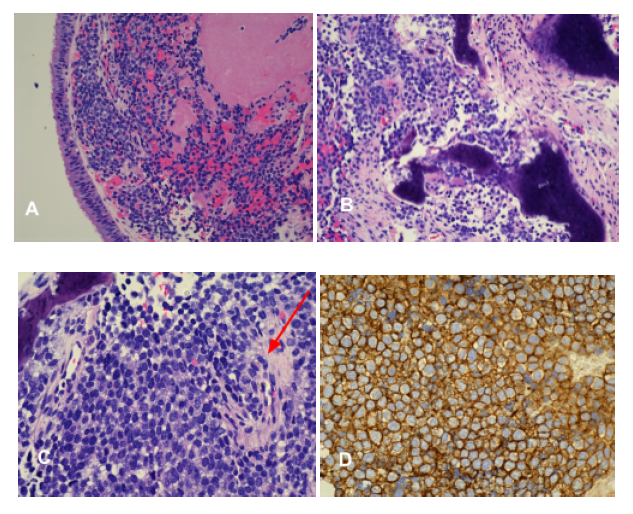
Figure 4: Multiple pathology slides demonstrating tumor invasion into and beneath the
submucosa (A) and (B). Small round blue cell tumor with few mitosis (red arrow) (C). CD99
sensitive marker for Ewing’s sarcoma showing diffuse membranous staining (D).
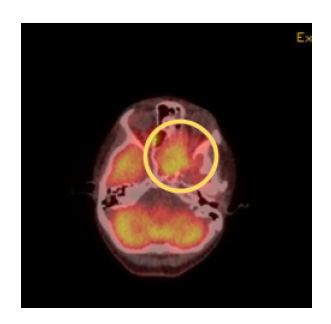
Figure 5: PET Whole Body Attenuation Correction demonstrating increased radiopharmaceutical activity (yellow circle) in the left sphenoid mass with extension of activity into the medial wall of the right orbit suggesting extension of the mass crossing the midline.
Discussion
Ewing Sarcoma (ES) is in a highly malignant “small, round cell” tumor family that, on
histology, presents with a higher nuclear to cytoplasmic ratio with indistinct cell membranes
with few mitosis. Additional members of this family include chondrosarcoma,
esthesioneuroblastoma, and other poorly differentiated tumors1. Incidence of ES is low in the
adult population and generally occurs in childhood or early adolescence period. ES typically
presents in the long bones of the axial skeleton or sacrum and ilium. Cases involving the skull
base only include 1-4% 3.
General imaging characteristics on radiograph include aggressive features and often
present with a permeative or moth-eaten osteolytic appearance in addition to cortical erosion
and periostitis. On MRI, the tumor features T1 hypo- to iso-intense signal with heterogeneous
enhancement on postcontrast imaging. The tumor also shows avid T2 hyperintense signal. 4
The differential considerations of these masses are broad but one consideration, being
skull based in location, is a chondrosarcoma. These tumors infrequently involve the craniofacial
region at around 2% of the time. Imaging features include a high T2 signal with vivid contrast
enhancement. On CT, chondrosarcoma demonstrates matrix calcification in 94% of cases.
Chondrosarcoma also typically presents in an “off-midline” location. 5 Another differential
consideration for this location is esthesioneuroblastoma, which often involves the ethmoid
sinus and extends through the lamina papyracea. The imaging findings of
esthesioneuroblastoma includes a lobulated mass with a T2-hyperintense cystic mass that
shows intralesional flow voids. The tumor also shows an avid enhancement.
Esthesioneuroblastoma has a bimodal distribution with one peak in young adults (around 20s)
and another peak in the 50-60 age group. A common presenting symptom is epistaxis.6
Treatment of choice is systemic chemotherapy with surgery and/or radiation with
chemotherapy depending on the location and size of tumor.6 Prognosis is poor depending on
size, site of primary tumor, tumor metastasis, and age of the patient. The 5-year survival rate is
approximately 50-75%. 2
Conclusion
Our patient in this case report presents with an atypical location of a highly malignant
pathology. Diagnosis is often delayed due to vague and non-specific symptoms. It is important
to know the imaging characteristics of Ewing sarcoma in order to make an accurate and efficient
diagnosis. This is a rare but aggressive tumor and providing appropriate therapy for the patient
in a timely manner is necessary to increase survival rates. Diagnosis and treatment of Ewing
sarcoma involve a multidisciplinary approach. In this case alone, ENT, Ophthalmology, Radiology, and Pathology were all required for the diagnosis and treatment of the patient. Treatment will require many of these same medical subspecialties in addition to radiation and chemotherapy treatments.
References
1. Freda PU, Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin
North Am. 1999;28(1):81-117, vi.
2. Murphey MD, Senchak LT, Mambalam PK, Logie CI, Klassen-Fischer MK,
Kransdorf MJ. From the radiologic pathology archives: ewing sarcoma family of
tumors: radiologic-pathologic correlation. Radiographics. 2013;33(3):803-831.
3. Aldandan A, Almomen A, Alkhatib A, Alazzeh G. Pediatrics Ewing’s Sarcoma of the
Sinonasal Tract: A Case Report and Literature Review. Case Rep Pathol.
2019; 2019:8201674.
4. Frazier AA. Radiologic Patterns of Ewing Sarcoma. Radiographics.
2013;33(3):802-802.
5. Varma DG, Ayala AG, Carrasco CH, Guo SQ, Kumar R, Edeiken J.
Chondrosarcoma: MR imaging with pathologic correlation. Radiographics.
1992;12(4):687-704.
6. Morita A, Ebersold MJ, Olsen KD, Foote RL, Lewis JE, Quast LM.
Esthesioneuroblastoma: prognosis and management. Neurosurgery.
1993;32(5):706-714; discussion 714-715.