Urticarial vasculitis: An unusual manifestation of ‘rhupus’
Prashant Kaushik, M.D., Chief, Division of Rheumatology, Northeastern Health System (NHS), Tahlequah, OK; Clinical Associate Professor of Medicine, Oklahoma State University College of Osteopathic Medicine Center for Health Sciences (OSU CHS) at the Cherokee Nation, Tahlequah, OK. Associate Program Director, Internal Medicine Residency Program, TMG/OMECO
Stephen Bastible, D.O., Resident, TMG/OMECO Internal Medicine Residency Program, Department of Internal Medicine, NHS, Tahlequah, OK
Matthew W. Gibson B.S., Matthew W. Gibson, MS III, OSU CHS, Tulsa, OK
Jana Baker, D.O., Associate Professor, OSU CHS; previous Program Director of the TMG/OMECO Internal Medicine Residency Program; Division of Hospitalist Medicine at the Eastern Oklahoma VA Health Care System
Guarantor of Submission: The corresponding author is the guarantor of submission.
Keywords: rhupus, rheumatoid arthritis, systemic lupus erythematosus, urticarial vasculitis
CONFLICT OF INTEREST
The authors have no financial or conflicts of interest to disclose.
Abbreviations: rheumatoid arthritis (RA), urticarial vasculitis (UV), hydroxychloroquine (HCQ), systemic lupus erythematosus (SLE), cutaneous lupus erythematosus (CLE), non-steroidal anti-inflammatory drugs (NSAIDs), methotrexate (MTX), disease-modifying antirheumatic drug (DMARD)
Abstract
A 43-year-old female presented to the clinic for rheumatological evaluation for chronic worsening joint pain and associated morning stiffness with family history of rheumatoid arthritis (RA) in her father and a daughter who is HLA-B27 positive. After initial clinic presentation, the patient was hospitalized for acute onset of a truncal skin rash which after biopsy was identified as being secondary to urticarial vasculitis (UV). Management consisted of oral methotrexate with discontinuation of previously initiated hydroxychloroquine (HCQ). While UV alone is not pathognomonic for lupus, patient-presentation both clinically and laboratory-wise favored systemic lupus erythematosus (SLE) (especially with presence of hypocomplementemia for C4) along with early rheumatoid arthritis(14.3.3η positivity), an overlapping condition also known as ‘rhupus’.
Introduction
Rhupus is a rare rheumatological condition with an estimated prevalence of 0.09% marked by an overlap in symptoms between RA and SLE1. The condition was first described by Toone in 1960 among a panel of 15 patients2, 3. As rhupus does not have a defined clinical classification/diagnostic criteria of its own, the condition relies on existing American College of Rheumatology criteria developed to assess for RA and SLE individually and then combining the clinico-laboratory findings in the perspective4,5.
As a systemic condition, SLE is known for a wide-ranging spectrum of clinical manifestations which can occur in virtually any system of the body. Dermatological manifestations, or cutaneous lupus erythematosus (CLE), may manifest as discoid skin changes, malar rash, and lupus tumidus, profundus, and panniculitis6. Additionally, there are other skin manifestations which can be associated with SLE. One such example includes urticarial vasculitis (UV) which is characterized as a vasculitis of the small vessels with characteristic skin involvement notable for lesions consisting of wheals which feature a burning sensation more often than itching and often result in a hyperpigmentation of the skin upon rash resolution, classical histology demonstrating leukocytoclastic vasculitis, with perivascular neutrophilic infiltrate7. While UV can be idiopathic, it is also known to result from underlying infection, drug reaction, paraneoplastic syndrome, or autoimmune disorder; commonly idiopathic etiology including an elevation of acute phase reactants with decreased serum complement on laboratory analysis8. UV is known to be a rare manifestation of SLE, estimated to affect 2% of patients with SLE9.
Case Presentation
History of Present Illness
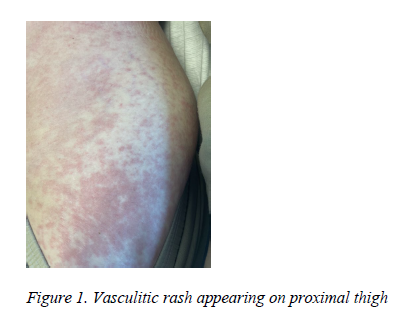
A 43-year-old female presented to the clinic on referral for rheumatological evaluation for joint pain which the patient noted had been present since age 27. The patient reported a family history of RA in her father and a daughter who is HLA-B27 positive with a possibility of axial spondyloarthritis. The patient described experiencing morning stiffness with varying duration. Joint pain was most severe in hands, wrists, elbows, and knees with a popping sensation experienced during ambulation. The patient had previously utilized non-steroidal anti-inflammatory drugs (NSAIDs) intermittently for symptomatic management. However, she noted that recently her joint pain had increased in severity with a decrease in effectiveness of the NSAIDs in reducing her pain. Subsequent to the initial clinic visit, the patient was hospitalized after breaking out in a malar rash, which was followed by the eruption of a hive-like rash which encompassed the majority of her trunk, abdomen, and extremities. (Figure 1) The patient remarked that the rash had been preceded for a short time by intermittent periods of cramping abdominal pain.
Laboratory and Pathology Studies
The patient was initially evaluated for inflammatory polyarthritis with further laboratory investigation conducted which subsequently revealed elevation in serum creatinine (0.9 mg/dL; normal range 0-0.8), decreased C4 (14; normal range:15-57 ), elevated ESR (16 mm/hr), positive 14.3.3η (normal is <0.2), with negative antibodies for ANA, CCP, RF, Scl-70, centromere, SSA, SSB, histone, Smith, Jo-1, hepatitis C, with negative hepatitis B surface antigen and a negative HLA B-27. Pathological analysis of punch biopsy of the rash while patient was in hospital was significant for "mild superficial perivascular lymphoid infiltrate with a few scattered neutrophils and eosinophils; focal vacuolar change along the dermal-epidermal junction. GMS negative for fungus. Review of mast cell tryptase immunostain failed to reveal any significant increase in the number of mast cells." Immunofluorescence could not be performed. A working diagnosis of UV was agreed upon by the treating hospitalist, pathologist and rheumatologist.
Management
Initial treatment initiated on the second clinic visit included oral hydroxychloroquine (HCQ) at a weight-based 200 mg daily for ‘rhupus’. However, the patient was concerned that the rash might have been due to the HCQ and she discontinued its use during hospitalization and upon discharge was prescribed a course of oral prednisone which was also eventually discontinued due to patient-reported feelings of agitation and an increase in appetite. On post-hospitalization follow-up evaluation in the rheumatology clinic, the patient, through shared decision-making,elected to proceed with discontinuation HCQ in favor of initiation of oral methotrexate (MTX) as a DMARD, with daily folic acid supplementation.
Discussion
The patient's clinical presentation in clinic and subsequent hospitalization in combination with laboratory and pathology studies is suggestive of early RA in addition to new-onset lupus in an overlapping format. One of the differentiating factors between UV and urticaria involves lesion-symptomatology. Typically, eruptions seen in UV are more prolonged in duration, frequently described as being painful rather than pruritic or, if elements of pruritus are present, they are more often described as painfully so, with resolution of the lesions leaving behind areas of residual hyperpigmentation.
UV is subdivided into 2 categories with the first being characterized by normal serum complement levels and with the second notable for diminished complement levels and increased levels of anti-C1q autoantibody, which serve as a diagnostic biomarker for hypocomplementemic UV. It has been observed that increased levels of anti-C1q are detected in up to 30% of patients with concomitant SLE and up to 80% of patients with concomitant SLE in the presence of glomerulonephritis. However, while UV without diminished complement is not known to be strongly associated with SLE, as many as 54% of patients with hypocomplementemic UV are associated with SLE, which has led to UV recognition as a potential harbinger of underlying systemic autoimmune disease such as SLE or Sjogren’s syndrome10. Among severe UV with systemic involvement, musculoskeletal, renal, gastrointestinal, pulmonary, and ocular systems
can be affected11. Regarding treatment, UV secondary to drug administration generally resolves after discontinuation of the offending agent; and for autoimmune etiologies, treatment often includes immunomodulation or immunosuppression with agents such as corticosteroids, hydroxychloroquine, methotrexate, novel biologic agents such as omalizumab, or plasmapheresis12.
Clinical Bottom Line
While not common, SLE and RA may appear concomitantly in an overlapping condition known as ‘rhupus’, which is well described in literature yet needs to be revived in daily clinical practice.
Urticarial vasculitis is marked by vasculitis of the small vessels with characteristic skin involvement consisting of wheals often accompanied by a burning sensation and often resulting in a hyperpigmentation of the skin upon resolution. Classical histology for UV includes leukocytoclastic vasculitis with perivascular neutrophilic infiltrate.
References
1. Panush RS, Edwards NL, Longley S, Webster E. 'Rhupus' syndrome. Arch Intern Med. 1988;148(7):1633-1636.
2. Toone EC Jr, Irby R, Pierce EL. The L.E. cell in rheumatoid arthritis. Am J Med Sci. 1960;240:599-608.
3. Antonini L, Le Mauff B, Marcelli C, Aouba A, de Boysson H. Rhupus: a systematic literature review. Autoimmun Rev. 2020;19(9):102612. doi:10.1016/j.autrev.2020.102612
4. Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative [published correction appears in Ann Rheum Dis. 2010 Oct;69(10):1892]. Ann Rheum Dis. 2010;69(9):1580-1588. doi:10.1136/ard.2010.138461
5. Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019;71(9):1400-1412. doi:10.1002/art.40930
6. Fairley JL, Oon S, Saracino AM, Nikpour M. Management of cutaneous manifestations of lupus erythematosus: A systematic review. Semin Arthritis Rheum. 2020;50(1):95-127. doi:10.1016/j.semarthrit.2019.07.010
7. Marzano AV, Tavecchio S, Venturini M, Sala R, Calzavara-Pinton P, Gattorno M. Urticarial vasculitis and urticarial autoinflammatory syndromes. G Ital Dermatol Venereol. 2015;150(1):41-50.
8. Venzor J, Lee WL, Huston DP. Urticarial vasculitis. Clin Rev Allergy Immunol. 2002;23(2):201-216. doi:10.1385/CRIAI:23:2:201
9. Diplomatico M, Gicchino MF, Ametrano O, Marzuillo P, Olivieri AN. A case of urticarial vasculitis in a female patient with lupus: Mycoplasma pneumoniae infection or lupus reactivation?. Rheumatol Int. 2017;37(5):837-840. doi:10.1007/s00296-016-3626-9
10. Her MY, Song JY, Kim DY. Hypocomplementemic urticarial vasculitis in systemic lupus erythematosus. J Korean Med Sci. 2009;24(1):184-186. doi:10.3346/jkms.2009.24.1.184
11. Gu SL, Jorizzo JL. Urticarial vasculitis. Int J Womens Dermatol. 2021;7(3):290-297. Published 2021 Jan 29. doi:10.1016/j.ijwd.2021.01.021
12. Kolkhir P, Grakhova M, Bonnekoh H, Krause K, Maurer M. Treatment of urticarial vasculitis: A systematic review. J Allergy Clin Immunol. 2019;143(2):458-466. doi:10.1016/j.jaci.2018.09.007