THE ACUTE EFFECTS OF β-FUNALTREXAMINE ON LIPOPOLYSACCHARIDE-INDUCED INFLAMMATION IN BV2 MURINE MICROGLIAL CELLS
Karissa Hodge, Oklahoma State University Center for Health Sciences
Daniel J. Buck, Oklahoma State University Center for Health Sciences
Subhas Das, Oklahoma State University Center for Health Sciences
Randall L. Davis, Oklahoma State University Center for Health Sciences
Corresponding Author: Randall L. Davis, randall.davis@okstate.edu
Oklahoma State University Center for Health Sciences
1111 West 17th Street, Tulsa, Oklahoma 74107
Conflict of Interest Statement:
The authors declare that there are no conflicts of interest.
Funding Sources:
This study was funded in part by Oklahoma Center for Advancement of Science & Technology Health Research Program-HR 18-033 (RLD) and the Oklahoma State University Center for Health Sciences, Office of the Vice President for Research (RLD).
Abstract
Inflammation is a crucial and driving factor in many pathological conditions. Additionally, neuroinflammation has been associated with poorer outcomes in brain and spinal cord injuries, as well as fueling and exacerbating neurodegenerative and neuropsychiatric conditions. Microglia are keystones of neuroinflammation, but efforts to isolate their role in neuroinflammation have been challenging due to heterogeneity based on brain region, sex, age, and/or disease. There is a dearth of pharmaceuticals that can cross the blood-brain barrier to target neuroinflammation without dependency or toxicity issues. Our lab has been investigating the impact of a well-tolerated, non-addictive irreversible μ-opioid antagonist and reversible kappa-opioid agonist, β-funaltrexamine (β-FNA), on neuroinflammation, peripheral inflammation, and inflammation-induced behavior deficits. Previously, our lab discovered that β-FNA has anti-inflammatory effects on normal human astrocytes and male C57BL/6J mice, as well as ameliorating effects on anxiety-like and sickness-like behavior in male C57BL/6J mice. Now, we examined the effects of acute β-FNA treatment on lipopolysaccharide (LPS)-stimulated BV2 murine microglial cells. We discovered that β-FNA dampens morphological changes associated with LPS-driven microglial activation and inhibits CCL2. Consequently, these findings provide novel insights into the cell-specific anti-inflammatory effects of β-FNA and its potential as a therapeutic agent.
Keywords: neuroinflammation, microglia, β-FNA, inflammation, chemokine, CCL2, CXL10, STAT1, BV2
1. Introduction
Neuroinflammation underlies and spurs brain and spinal cord inflammation, neurodegenerative conditions, and neuropsychiatric conditions, such as schizophrenia and depression 1–6. Microglia, the resident macrophages of the central nervous system, act as sentinels and drive inflammation by triggering the immune system when a threat is perceived 7–13. While research has shed light on the role of microglia in driving neuroinflammation, the heterogeneity of these cells based on sex, brain region, age and/or disease poses challenges in determining effects 11,12,14–19. Combined with the impermeability of the blood-brain barrier, it is often challenging to develop pharmaceuticals that can access the central nervous system without being toxic or causing dependency.
Previously, the authors discovered that β-FNA, an irreversible μ-opioid antagonist and reversible kappa opioid agonist, is a potent non-toxic, non-addictive anti-inflammatory agent that can pass the blood-brain barrier and robustly decrease neuroinflammation 20–25. Interestingly, it was discovered that many of its anti-inflammatory actions involve modulation of non-opioid receptor targets, such as NF-κB and TLR4 22,23,25,26. To date, in vitro studies have demonstrated that β-FNA selectively reduces CXCL10, but not CCL2 or IL-6, via inhibition of NF-κB p50 and p65 subunit phosphorylation and p38 MAPK phosphorylation in stimulated normal human astrocytes 22,23. Additionally, C20 human microglial cells exhibited no sensitivity to the anti-inflammatory actions of acute β-FNA 25. The authors also discovered through in vivo studies with C57BL/6J mice that β-FNA reduces anxiety- and sickness-like behavior and inhibition of cytokine/chemokine expression in the brain and peripheral tissues20,21,24.
The purpose of this study was to further elucidate the effects of acute β-FNA treatment on LPS-induced neuroinflammation by investigating its effects on BV2 murine microglial cells via measurement of the inflammatory chemokines CCL2 and CXCL10, as well as the upstream activator STAT1. Furthermore, drug cytotoxicity and cell morphological changes under control and experimental conditions were examined.
2. Methodology
2.1. Cell Culture
BV2 murine microglial cells (cat# AMS.EP-CL-0493, AMSBIO, Cambridge, MA) were cultured using reagents and procedures provided by the supplier. Cultures were maintained in a humidified incubator at 37°C, 5% CO2, and 95% air in growth media (Dulbecco’s Modified Eagle Medium, cat#10-013-CV, Corning) supplemented with 2.5 mmol/L L-glutamine (cat#25030-081, Gibco), 10% fetal bovine serum (FBS; cat#26140-079, Gibco), and 1% penicillin/streptomycin. Culture medium was replenished every 48h, and experimental cultures were switched to a low-serum media (1% FBS) 24h prior to stimulation.
2.2. LDH Cytotoxicity Assay
A standard LDH cytotoxicity assay kit (cat#88954, Thermo Scientific) was used according to manufacturer’s instructions to determine cytotoxicity associated with various concentrations of LPS, β-FNA, and LPS plus β-FNA. Per the International Organization for Standardization (ISO), cell toxicity was defined as greater than 30% cell death (cell viability < 70%) 27.
2.3. Experimental Protocol
BV2 murine microglial cells were seeded and maintained in growth media until 80-90% confluent at the time of stimulation. Cells designated for Western blot assays were seeded in 6-well plates at a density of 1.5×105 cells/well, while cells designated for ELISA assays were seeded in 24-well plates at a density of 3.0x104 cells/well. Cells were co-exposed in growth medium to LPS (4μg/mL) or saline and β-FNA (3μM) or saline. (Drug concentrations and duration of stimulation were based on findings from previous experiments.) Cell culture medium or whole cell lysate was collected based upon the assay being performed.
2.4. Enzyme-linked Immunosorbent Assay (ELISA)
Standard dual-antibody solid phase immunoassays (ELISA Development Kit; Peprotech, Rocky Hill, NJ) were used according to manufacturer’s instructions for quantitation of secreted CXCL10 (cat#900-K153) and CCL2 (cat#900-K126) into BV2 cell culture medium. Absorbance was read at 405 nm (650 nm wavelength correction) using a BIOTEK Synergy 2 Multi-Detection Microplate Reader and quantified using BioTek Gen5 software. Levels were normalized to total cell protein as determined by bicinchoninic (BCA) protein assay.
2.5. Immunoblotting Assay (Western Blot)
Western blot analysis was used to measure the expression of phosphorylated STAT1 (p-STAT1) and STAT1 in BV2 whole cell lysates. Thirty micrograms of total protein were loaded on a 7.5% polyacrylamide gel (cat#1610171, Bio-Rad), electrophoresed, and then transferred to a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked in tris-buffered saline containing 0.1% Tween (TBST) and 5% bovine serum albumin (BSA, cat#BP1605-100, Fisher) for 2h and then incubated overnight at 4°C in 1:1000 target-specific 1° antibody (p-STAT1, cat#9171; STAT1, cat#14995; β-tubulin, cat#2146S, Cell Signaling Technology). Membranes were then washed in TBST (3 x 20m), incubated at room temperature for 2h in 2° antibody (1:10,000 Goat anti-Rabbit IgG in TBST with 0.2% SDS; cat#925-32211, Li-Cor), and washed again (2 × 20m TBST, 2 × 10m TBS). A Li-Cor CLx Odyssey Infrared Imaging System was used to image and quantify relative protein signals using direct detection of secondary antibodies with near-infrared fluorescent dyes. Stripping buffer (Cat# 21059; Thermo Fisher Scientific) was utilized to re-probe membranes with subsequent primary antibodies for the purposes of data normalization. β-tubulin was used as the loading control for normalization of total protein levels, while total protein levels were used to normalize phosphorylated protein levels. Results were presented relative to the control group.
2.6 Statistical Analysis
Data were analyzed using two-way ANOVA (β-FNA × LPS) and Fisher’s LSD for pairwise comparisons. Data are presented as mean ± SEM, and p-values < 0.05 are considered statistically significant. GraphPad Prism 10.0.1 software (GraphPad Inc, San Diego, CA) was used for data analysis and figure preparation.
3. Results
3.1. Cytotoxicity of LPS stimulation and/or β-FNA treatment
BV2 cells were treated with a serial dilution of LPS concentrations (0 μg/mL to 300 μg/mL). None of the tested concentrations reduced cell viability below 70%, and the mean viability for each concentration ranged from 86-93% (Fig. 1A). BV2 cells were also treated with a serial dilution of β-FNA (0 μM to 90 μM) to determine if treatment with β-FNA alone was cytotoxic. None of the tested concentrations reduced cell viability below 70%, and the mean viability for each concentration ranged from 90-99% (Fig. 1B). A final assay was performed to determine if there was a concentration of β-FNA that could induce cytotoxicity in BV2 cells when stimulated with 4 μg/mL LPS. BV2 cells were stimulated with 4 μg/mL and then immediately treated with a serial dilution of β-FNA (0 μM to 90 μM). None of the tested concentrations reduced cell viability below 70%, and the mean viability for each concentration ranged from 89-94% (Fig. 1C). These results indicate that BV2 cells are resilient to very high concentrations of LPS and/or β-FNA, and the concentrations of LPS and β-FNA utilized in subsequent experiments would not induce cytotoxicity.
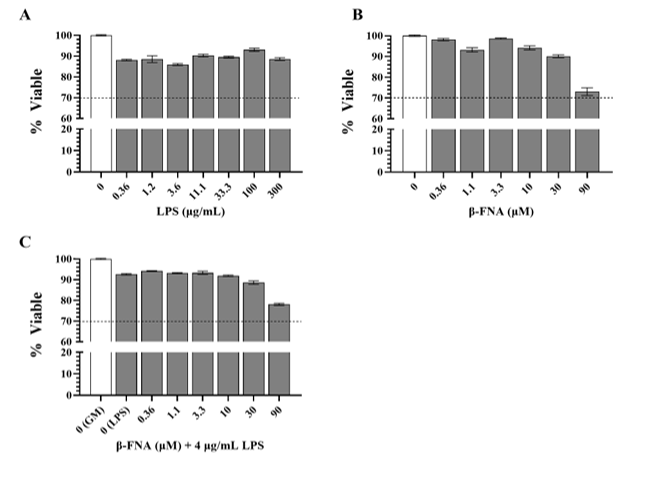
Fig. 1: Cytotoxicity of multiple concentrations of LPS, β-FNA, and β-FNA + LPS. Standard LDH cytotoxicity assays were used to determine if (A) LPS (n=7-12/concentration), (B) β-FNA (n=8-12/concentration), and (C) β-FNA + LPS (n=12-17/concentration) induced cell cytotoxicity (< 70% viability). Concentrations ranged from 0-300 μg/mL for LPS and 0-90 μM for β-FNA. For β-FNA + LPS, concentrations for β-FNA varied from 0-90 μM, but LPS was held constant at 4 μg/mL. Data are presented as mean ± SEM. The dashed line indicates the threshold of cytotoxicity; any data bars that fell below this line indicated a cytotoxic concentration.
3.2. Effects of β-FNA on LPS-driven rises in pro-inflammatory chemokines
ELISA assays were performed on BV2 cell culture medium to determine whether β-FNA treatment affected LPS-driven elevations in CCL2 and CXCL10. Two-way ANOVA also revealed that there was a significant main effect of LPS (F1, 65 = 1492.00, p < 0.0001) and a significant interaction of main effects (F1, 65 = 5.21, p < 0.05) on CCL2 levels in BV2 cells (Fig. 2A). While β-FNA trended towards a main effect on CCL2 levels, it was not significant (F1, 65 = 3.96, p = 0.051). Pairwise comparisons revealed that both the LPS and β-FNA + LPS groups had higher levels of CCL2 than the saline or β-FNA groups, which were similar (p < 0.0001 and p = 0.83, respectively). Additionally, the β-FNA+LPS group had lower levels of CCL2 than the LPS group (p < 0.005). These results indicate that β-FNA attenuates LPS-induced elevations in CCL2.
Two-way ANOVA revealed a significant main effect of LPS (F1, 64 = 841.40, p < 0.0001), but no significant main effect of β-FNA (F1, 64 = 0.14, p = 0.71) or interaction of main effects (F1, 64 = 0.52, p = 0.47) on CXCL10 levels in BV2 cells (Fig. 2B). Pairwise comparisons revealed that the saline and β-FNA groups were similar (p = 0.80), as well as the LPS and β-FNA + LPS groups (p = 0.46). Additionally, the LPS and β-FNA+LPS groups had significantly higher levels of CXCL10 than the saline and β-FNA groups (p < 0.0001). These results indicate that β-FNA does not alter CXCL10 levels in BV2 cells.
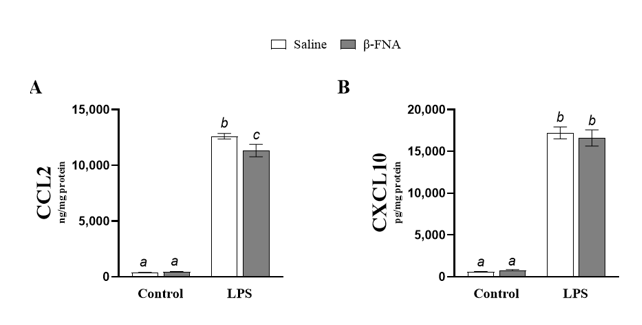
Fig. 2: Acute effects of β-FNA on LPS-induced elevations of CCL2 and CXCL10 in BV2 cells. BV2 cells were stimulated with 4 μg/mL LPS or control and then immediately dosed with either 3μM B-FNA or saline. Cells were harvested 24h after stimulation, and (A) CCL2 and (B) CXCL10 were quantified in cell supernatant by ELISA and normalized to protein (as determined by BCA assay utilizing whole cell lysates). Data are presented as mean ± SEM. (A) Two-way ANOVA indicated a significant main effect of LPS (p < 0.0001) and a significant interaction of main effects (p < 0.05), but no significant effect of β-FNA (p = 0.051) on CCL2 levels in BV2 cells (n=16-18/group). (B) Two-way ANOVA also indicated a significant main effect of LPS (p < 0.0001), but no significant main effect of β-FNA (p = 0.71) or interaction of main effects (p = 0.49) on CXCL10 levels in BV2 cells (n=15-18/group). Pairwise comparisons were assessed using Fisher's LSD test; bars with letters in common indicate data are not significantly different (p > 0.05).
3.3. Effects of β-FNA on STAT1 pathway in LPS-stimulated BV2 cells
Two-way ANOVA revealed a significant main effect of LPS (F1, 55 = 153.2, p < 0.0001), but no main effect of β-FNA (F1, 55 = 0.17, p =0.68) or interaction of main effects on p-STAT1 levels in BV2 cells (F1, 55 = 1.30, p =0.26). Pairwise comparisons showed that saline and β-FNA groups were similar (p = 0.62), as were LPS and β-FNA + LPS groups (p = 0.26) (Fig. 3). Both LPS and β-FNA + LPS groups had significantly higher levels of p-STAT1 than either saline or β-FNA groups (p < 0.0001). Collectively, these results indicate that β-FNA does not significantly affect p-STAT1 levels in BV2 cells.
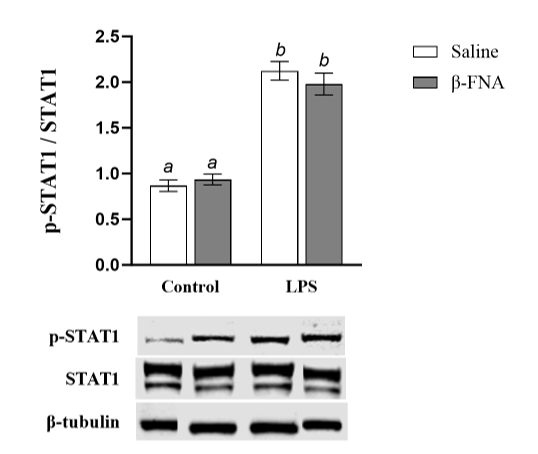
Fig. 3: Acute effects of β-FNA on LPS-induced activation of STAT1 in BV2 cells. BV2 cells were stimulated with 4 μg/mL LPS or control and then immediately dosed with either 3μM β-FNA or saline. Cells were harvested 24h after stimulation. Levels of p-STAT1, STAT1, and β-tubulin were determined in whole cell lysate by Western Blot using 30μg total protein (as determined by BCA assay). Levels of p-STAT1 were normalized to STAT-1 and STAT-1 was normalized to housekeeping β-tubulin. Data are presented as mean ± SEM. Two-way ANOVA indicated a significant main effect of LPS (p < 0.0001), no main effect of β-FNA (p = 0.68), and no significant interaction of main effects (p = 0.26) on p38 MAPK levels in BV2 cells (n=14-16/group). Pairwise comparisons were assessed using Fisher's LSD test; bars with letters in common indicate data are not significantly different (p > 0.05). Representative Western blots are shown below the graph.
3.4. Morphological changes in BV2 cells from LPS and/or β-FNA treatment
Microglia alternate between an M1 or M2 phenotype based on the activating stimulus 10,28. LPS or IFN-γ stimulates microglia to convert to the pro-inflammatory and neurotoxic M1 phenotype, characterized by an amoeboid cell body with retracted cell processes 10,11,28,29. On the other hand, anti-inflammatory cytokines (IL-4, IL-13, etc.) stimulate microglia to convert to the anti-inflammatory and neuroprotective M2 phenotype, characterized by an elongated cell body with unipolar or bipolar processes 10,11,28,29. The ability to convert between phenotypes creates a bidirectional continuum of morphological changes, where microglia can present with aspects of both phenotypes while converting from one phenotype to the other 10,11,30.
Pictures of BV2 experimental cultures were taken 24h after LPS stimulation to illustrate morphological differences between treatment groups. Cells treated with a saline control or β-FNA alone exhibited a typical M2 phenotype (Figs. 4A, 4B). Conversely, cells treated with LPS exhibited a conversion to the M1-like phenotype, with complete loss of processes and a rounded, amoeboid cell body (Fig. 4C). Interestingly, when LPS-stimulated BV2 cells were treated with β-FNA, they demonstrated a “bushy” morphology (Fig. 4D). This intermediate phenotype was characterized by bloated, truncated processes and an enlarged cell body 10,31. Therefore, it appears that β-FNA mitigates LPS-induced morphological changes and hinders the complete conversion to the M1-like phenotype.
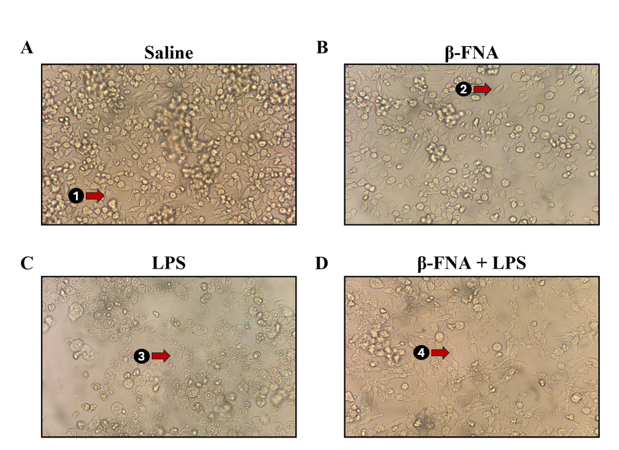
Fig. 4: BV2 morphology with LPS stimulation and/or β-FNA treatment. BV2 cells were seeded in 6-well plates at 3.0 × 105 cells per well and maintained in growth media at 37 °C and 5% CO2 until 90-95% confluent. Cells were starved 24h prior to stimulation and then stimulated with 4 μg/mL LPS or control and then immediately dosed with either 3μM β-FNA or saline. Representative pictures of (A) saline, (B) β-FNA, (C) LPS, and (D) β-FNA + LPS experimental cell cultures were taken 24h after LPS stimulation with an iPhone 14 camera aimed through a microscope ocular lens. Red arrows point to (1, 2) a resting ramified morphology, (3) an activated amoeboid morphology, and (4) a “bushy” intermediate morphology
4. Discussion
To the best of the authors’ knowledge, these studies are the first to examine the specific effects of acute β-FNA treatment on LPS-induced inflammation in BV2 cells. The study demonstrated that acute β-FNA treatment of LPS-induced inflammation in BV2 murine microglial cells results in selective inhibition of CCL2 and amelioration of inflammation-induced morphological changes. Specifically, β-FNA attenuates LPS-driven elevations in CCL2 but does not impact CXCL10 levels. Interestingly, previous work conducted by the authors has shown that β-FNA attenuates CXCL10 levels but not CCL2 levels in normal human astrocytes (NHA) in a MOR-independent manner when stimulated with IL-1β 22,23. Furthermore, the authors’ research has also shown that acute β-FNA treatment has sex-specific effects in an LPS-driven in vivo model of inflammation, in which brain levels of both chemokines were reduced 24h post-treatment in males, but not in females20,21,24. Consequently, there appears to be disparities in responses to β-FNA between cell culture and animal models.
TLR4 activation can lead to different inflammatory responses based upon whether the MyD88-dependent or MyD88-independent pathway is triggered. When LPS binds to TLR4, the adaptor protein MyD88 is recruited and initiates a cascade of downstream effects that trigger the inhibitor of nuclear factor-κB kinase (IKK) complex and the mitogen-activated protein kinases (MAPKs) to activate the transcription factors NF-κB and AP-1, respectively 32. This upregulates pro-inflammatory gene expression and induces immune cell and inflammasome activation, pro-inflammatory cytokine production, and regulation of development, growth, and apoptosis32. However, upon endocytosis, TLR4 can then bind a second set of adaptor proteins, TRAM and TRIF, within the endosome to initiate MyD88-independent signaling 32. MyD88-independent signaling results in alternative activation of NF-κB and AP-1, as well activation of the IRF3 and IRF7 transcription factors, which are responsible for inducing type I IFN cytokines that activate intracellular antimicrobial programs, influence immune responses, and upregulate other important pro-inflammatory mediators, such as the chemokine CXCL10 (IP-10) 33–36. Furthermore, the type I IFN IFN-β can act in an autocrine fashion to bind to the IFN-α/β receptor (IFNAR), triggering the formation of STAT1 homodimers and STAT1/STAT2 heterodimers that directly bind to gamma interferon activation sites (GAS) on DNA 33–36. This triggers the release of the type II IFN IFN-γ, which further amplifies pro-inflammatory gene expression and activates antiviral defenses, increases antiproliferative activities, and stimulates adaptive immunity 33–36.
The discrepancy in CCL2 and CXCL10 inhibition between in vitro and in vivo studies may be due to differences in activation of the MyD88-dependent or MyD88-independent pathways. Human monocytes and macrophages and mouse microglia and macrophages stimulated with LPS have been shown to regulate CCL2 in a TLR4 MyD88-dependent manner and CXCL10 in a TLR4 MyD88-independent manner 37–40. MAPK cascades, while directly targeted by MyD88-dependent pathways, can also be amplified indirectly through STAT1 homodimers induced by IFN-β, which is upregulated by the TLR4 TRAM-TRIF adaptors of the MyD88-independent pathway 32–36. Remarkably, it has also been shown that rodent astrocytes do not release IFN-β after LPS, but still regulate CXCL10 through tyrosine phosphorylation of STAT1 to form STAT1 homodimers 41,42. Furthermore, CXCL10 mRNA is predominantly associated with astrocytes during viral and nonmicrobial neuroinflammation and actively participates in microglial activation, suggesting that the in vivo effects may, in part, be due to inhibition of microglial activation via inhibition of CXCL10 in astrocytes 43,44. Additionally, it is possible that β-FNA may favorably inhibit MyD88-dependent and MyD88-indepent pathways in cell-specific ways, preferentially inhibiting MyD88-dependent pathways for microglia and MyD88-independent pathways and/or tyrosine kinases for astrocytes, resulting in the disparity of chemokine effects between cell types. Since it has been established that acute β-FNA treatment inhibits both CCL2 and CXCL10 in the brains and/or brain regions of male mice but not female mice, this indicates that the sex-specific cell differences, region-dependent responses, and/or the interplay of glial cells are crucial to the overall anti-inflammatory response of β-FNA.
The disparities in BV2 chemokine response may also be attributed to the concentration of β-FNA used during experimentation. Previous studies have demonstrated that inflammation in BV2 cells was not significantly inhibited until the concentration of β-FNA approached or exceeded 10 μM, and studies on the anti-inflammatory effects of β-FNA in neuron and glial mixed cultures were not apparent until β-FNA reached 30μM23,45. It is possible that lower β-FNA concentrations may have chemokine specificity, which is abolished with higher concentrations. In the future, increasing the concentration of β-FNA may parse out additional anti-inflammatory effects.
It has been well-documented that microglia display heterogeneity in density, phenotype, function, and/or transcriptomic profile based on brain region, age, and/or sex 11,12,14–17. As such, the brain region from which the microglial cell line was derived could significantly alter interpretations of a study’s findings. Additionally, the preponderance of experimental research has been performed on male cell lines and animals, and, therefore, has driven an unbalanced and androcentric approach to pharmaceutical development and disease treatment 46. The importance of this cannot be overstated, as evidence continues to mount that female inflammatory, immune, drug, and pain responses are often significantly different, and sometimes antithetically opposed, to male responses 18,24,47–55.
BV2 cells were created in 1989 by immortalizing murine primary microglial cell cultures with a -raf/v-myc oncogene-carrying retrovirus (J2) 56. The cells were harvested from mixed brain cultures derived from 1-week-old female C57BL/6 mice and purified by shaking 56,57. Mice are usually classified as adults with mature brains at the age of eight weeks, and recent research has suggested that mouse brain maturation and changes may occur for onwards of six months 58,59. As such, BV2 experimental results could be affected by regional heterogeneity, passage-to-passage changes in component populations, sex-specific cell responses, and cell line sampling age. Consequently, this highlights the need for cell line validity, consistency, and specificity when comparing experimental results across studies.
β-FNA did not affect p-STAT1/STAT1 in LPS-stimulated BV2 cells. STAT1 phosphorylation drives CXCL10 expression; therefore, the study’s findings that β-FNA did not alter p-STAT1/STAT1 and CXCL10 levels support the hypothesis that β-FNA may predominantly affect the MyD88-dependent pathway in BV2 microglia. Interestingly, Shen et al. has shown that BV2 cells predominantly regulate CXCL10 through MAPK pathways; however, the BV2 cells were co-stimulated with IFN-γ and incubated for a period of 48 hours at a concentration of 100ng/mL LPS 60. The conflict in our results and this study suggest that β-FNA may not affect p38 MAPK, which would imply that β-FNA inhibits the MyD88-dependent pathway further downstream, or that β-FNA has concentration-dependent effects. Additionally, it is possible that co-stimulation, stimulation duration, and/or LPS concentration may differentially affect production pathways of CXCL10 in BV2 cells.
While the effects of β-FNA on LPS-stimulated BV2 cells were limited to inhibiting CCL2, it is worth mentioning that β-FNA had a noticeable anti-inflammatory effect on BV2 morphology. The LPS-only cells switched to a de-ramified amoeboid body, which is indicative of microglial activation and a M1 phenotype 10,29. With β-FNA treatment, the cells exhibited an intermediate phenotype characterized by bloated, truncated processes and an enlarged cell body. This indicates that β-FNA ameliorates LPS-induced morphological changes and opposes activation. As such, further studies are needed to further elucidate the effects of β-FNA on microglial activation and neuroinflammation.
5. Conclusions
In summary, these studies are the first to the authors’ knowledge that examine the specific, acute effects of β-FNA on LPS-stimulated BV2 cells. It was found that β-FNA ameliorated microglial activation and inhibited CCL2 expression. β-FNA did not affect CXCL10 or p-STAT1/STAT1 expression; however, sex, brain region, and sampling age of the BV2 cell line made it difficult to determine if other factors may be obscuring potential effects. Future efforts should focus on expanding experiments to look deeper into signaling pathways and crosstalk to further parse out the cell and sex-specific effects of β-FNA on MyD88-dependent and MyD88-independent pathways. Regardless, β-FNA continues to show therapeutic potential for reducing neuroinflammation and mitigating microglial activation.
Appendix A: List of Abbreviations
ANOVA: Analysis of Variance
β-FNA: beta-funaltrexamine
BCA: bicinchoninic assay
CCL2 : C-C motif chemokine ligand 2
CXCL10: C-X-C motif chemokine 10
CO2: carbon dioxide
ELISA: Enzyme-Linked Immunosorbent Assay
FBS: fetal bovine serum
IFN-β: interferon-beta
IFN-γ: interferon-gamma
IL-1: interleukin-1
IL-4: interleukin-4
IL-6: interleukin-6
IL-13: interleukin-13
LDH: lactate dehydrogenase
LPS: lipopolysaccharide
LSD: least significant difference
MyD88: myeloid differentiation primary response 88
NF-κB: nuclear factor kappa B
NHA: normal human astrocytes
p-STAT1: phosphorylated STAT1
p38 MAPK: p38 mitogen-activated protein kinase
SEM: standard error of mean
STAT1: signal transducer and activator of transcription 1
TBST: tris-buffered saline + Tween 20
TLR4: Toll-like receptor 4
TNF-α: tumor necrosis factor-alpha
REFERENCES
1. Müller N, Schwarz MJ. A psychoneuroimmunological perspective to Emil Kraepelins dichotomy: schizophrenia and major depression as inflammatory CNS disorders. Eur Arch Psychiatry Clin Neurosci. 2008;258 Suppl 2:97-106. doi:10.1007/s00406-008-2012-3
2. Bauer ME, Teixeira AL. Inflammation in psychiatric disorders: what comes first? Ann N Y Acad Sci. 2019;1437(1):57-67. doi:10.1111/nyas.13712
3. Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822-1832. doi:10.1038/s41591-019-0675-0
4. Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. 2014;79:1-12. doi:10.1016/j.neures.2013.10.004
5. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918-934. doi:10.1016/j.cell.2010.02.016
6. Berk M, Williams LJ, Jacka FN, et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013;11:200. doi:10.1186/1741-7015-11-200
7. Brandi E, Torres-Garcia L, Svanbergsson A, et al. Brain region-specific microglial and astrocytic activation in response to systemic lipopolysaccharides exposure. Front Aging Neurosci. 2022;14:910988. doi:10.3389/fnagi.2022.910988
8. Franco R, Lillo A, Rivas-Santisteban R, Reyes-Resina I, Navarro G. Microglial Adenosine Receptors: From Preconditioning to Modulating the M1/M2 Balance in Activated Cells. Cells. 2021;10(5). doi:10.3390/cells10051124
9. DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139 Suppl 2(Suppl 2):136-153. doi:10.1111/jnc.13607
10. Guo S, Wang H, Yin Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front Aging Neurosci. 2022;14:815347. doi:10.3389/fnagi.2022.815347
11. Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4(4):399-418. doi:10.1007/s11481-009-9164-4
12. Dadwal S, Heneka MT. Microglia heterogeneity in health and disease. FEBS Open Bio. Published online November 9, 2023. doi:10.1002/2211-5463.13735
13. Furube E, Kawai S, Inagaki H, Takagi S, Miyata S. Brain Region-dependent Heterogeneity and Dose-dependent Difference in Transient Microglia Population Increase during Lipopolysaccharide-induced Inflammation. Sci Rep. 2018;8(1):2203. doi:10.1038/s41598-018-20643-3
14. Tan YL, Yuan Y, Tian L. Microglial regional heterogeneity and its role in the brain. Mol Psychiatry. 2020;25(2):351-367. doi:10.1038/s41380-019-0609-8
15. Sun J, Song Y, Chen Z, et al. Heterogeneity and Molecular Markers for CNS Glial Cells Revealed by Single-Cell Transcriptomics. Cell Mol Neurobiol. 2022;42(8):2629-2642. doi:10.1007/s10571-021-01159-3
16. Spurgat MS, Tang SJ. Single-Cell RNA-Sequencing: Astrocyte and Microglial Heterogeneity in Health and Disease. Cells. 2022;11(13). doi:10.3390/cells11132021
17. Lee J, Kim SW, Kim KT. Region-Specific Characteristics of Astrocytes and Microglia: A Possible Involvement in Aging and Diseases. Cells. 2022;11(12). doi:10.3390/cells11121902
18. Gregus AM, Levine IS, Eddinger KA, Yaksh TL, Buczynski MW. Sex differences in neuroimmune and glial mechanisms of pain. Pain. 2021;162(8):2186-2200. doi:10.1097/j.pain.0000000000002215
19. Nissen JC. Microglial Function across the Spectrum of Age and Gender. Int J Mol Sci. 2017;18(3). doi:10.3390/ijms18030561
20. Davis RL, Stevens CW, Thomas Curtis J. The opioid antagonist, β-funaltrexamine, inhibits lipopolysaccharide-induced neuroinflammation and reduces sickness behavior in mice. Physiol Behav. 2017;173:52-60. doi:10.1016/j.physbeh.2017.01.037
21. Myers S, McCracken K, Buck DJ, Curtis JT, Davis RL. Anti-inflammatory actions of β-funaltrexamine in a mouse model of lipopolysaccharide-induced inflammation. Inflammopharmacology. 2023;31(1):349-358. doi:10.1007/s10787-022-01113-9
22. Davis RL, Das S, Thomas Curtis J, Stevens CW. The opioid antagonist, β-funaltrexamine, inhibits NF-κB signaling and chemokine expression in human astrocytes and in mice. Eur J Pharmacol. 2015;762:193-201. doi:10.1016/j.ejphar.2015.05.040
23. Davis RL, Das S, Buck DJ, Stevens CW. β-Funaltrexamine inhibits chemokine (CXCL10) expression in normal human astrocytes. Neurochemistry International. 2013;62(4):478-485. doi:10.1016/j.neuint.2013.01.013
24. Myers S, McCracken K, Buck DJ, Curtis JT, Davis RL. Anti-inflammatory effects of β-FNA are sex-dependent in a pre-clinical model of LPS-induced inflammation. J Inflamm . 2023;20(1):4. doi:10.1186/s12950-023-00328-z
25. Davis RL, McCracken K, Buck DJ. β-funaltrexamine differentially modulates chemokine and cytokine expression in normal human astrocytes and C20 human microglial cells. Neuroimmunology and. Published online 2020. https://nnjournal.net/article/view/3598
26. Stevens CW, Aravind S, Das S, Davis RL. Pharmacological characterization of LPS and opioid interactions at the toll-like receptor 4. Br J Pharmacol. 2013;168(6):1421-1429. doi:10.1111/bph.12028
27. Iso EN. 10993-5: 2009—Biological Evaluation of Medical Devices—Part 5: Tests for in vitro Cytotoxicity (ISO 10993-5: 2009). German Version.
28. Sakai A, Takasu K, Sawada M, Suzuki H. Hemokinin-1 gene expression is upregulated in microglia activated by lipopolysaccharide through NF-κB and p38 MAPK signaling pathways. PLoS One. 2012;7(2):e32268. doi:10.1371/journal.pone.0032268
29. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173(4):649-665. doi:10.1111/bph.13139
30. Jonas RA, Yuan TF, Liang YX, Jonas JB, Tay DKC, Ellis-Behnke RG. The spider effect: morphological and orienting classification of microglia in response to stimuli in vivo. PLoS One. 2012;7(2):e30763. doi:10.1371/journal.pone.0030763
31. Crews FT, Vetreno RP. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology . 2016;233(9):1543-1557. doi:10.1007/s00213-015-3906-1
32. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2021;78(4):1233-1261. doi:10.1007/s00018-020-03656-y
33. Zanin N, Viaris de Lesegno C, Lamaze C, Blouin CM. Interferon Receptor Trafficking and Signaling: Journey to the Cross Roads. Front Immunol. 2020;11:615603. doi:10.3389/fimmu.2020.615603
34. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18(9):545-558. doi:10.1038/s41577-018-0029-z
35. Negishi H, Taniguchi T, Yanai H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb Perspect Biol. 2018;10(11). doi:10.1101/cshperspect.a028423
36. Michalska A, Blaszczyk K, Wesoly J, Bluyssen HAR. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front Immunol. 2018;9:1135. doi:10.3389/fimmu.2018.01135
37. Dean JM, Wang X, Kaindl AM, et al. Microglial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microglia in vitro. Brain Behav Immun. 2010;24(5):776-783. doi:10.1016/j.bbi.2009.10.018
38. Bandow K, Kusuyama J, Shamoto M, Kakimoto K, Ohnishi T, Matsuguchi T. LPS-induced chemokine expression in both MyD88-dependent and -independent manners is regulated by Cot/Tpl2-ERK axis in macrophages. FEBS Lett. 2012;586(10):1540-1546. doi:10.1016/j.febslet.2012.04.018
39. Kawai T, Takeuchi O, Fujita T, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167(10):5887-5894. doi:10.4049/jimmunol.167.10.5887
40. Akhter N, Hasan A, Shenouda S, et al. TLR4/MyD88 -mediated CCL2 production by lipopolysaccharide (endotoxin): Implications for metabolic inflammation. J Diabetes Metab Disord. 2018;17(1):77-84. doi:10.1007/s40200-018-0341-y
41. Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59(2):242-255. doi:10.1002/glia.21094
42. Krasowska-Zoladek A, Banaszewska M, Kraszpulski M, Konat GW. Kinetics of inflammatory response of astrocytes induced by TLR 3 and TLR4 ligation. J Neurosci Res. 2007;85(1):205-212. doi:10.1002/jnr.21088
43. Clarner T, Janssen K, Nellessen L, et al. CXCL10 triggers early microglial activation in the cuprizone model. J Immunol. 2015;194(7):3400-3413. doi:10.4049/jimmunol.1401459
44. Phares TW, Stohlman SA, Hinton DR, Bergmann CC. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J Virol. 2013;87(6):3382-3392. doi:10.1128/JVI.03307-12
45. Wu CC, Chang CY, Shih KC, et al. β-Funaltrexamine Displayed Anti-inflammatory and Neuroprotective Effects in Cells and Rat Model of Stroke. Int J Mol Sci. 2020;21(11). doi:10.3390/ijms21113866
46. Sandberg K, Umans JG, Georgetown Consensus Conference Work Group. Recommendations concerning the new U.S. National Institutes of Health initiative to balance the sex of cells and animals in preclinical research. FASEB J. 2015;29(5):1646-1652. doi:10.1096/fj.14-269548
47. Martinez-Muniz GA, Wood SK. Sex Differences in the Inflammatory Consequences of Stress: Implications for Pharmacotherapy. J Pharmacol Exp Ther. 2020;375(1):161-174. doi:10.1124/jpet.120.266205
48. Seney ML, Glausier J, Sibille E. Large-Scale Transcriptomics Studies Provide Insight Into Sex Differences in Depression. Biol Psychiatry. 2022;91(1):14-24. doi:10.1016/j.biopsych.2020.12.025
49. Oi Yan Chan J, Moullet M, Williamson B, Arends RH, Pilla Reddy V. Harnessing Clinical Trial and Real-World Data Towards an Understanding of Sex Effects on Drug Pharmacokinetics, Pharmacodynamics and Efficacy. Front Pharmacol. 2022;13:874606. doi:10.3389/fphar.2022.874606
50. Carvalho Silva R, Pisanu C, Maffioletti E, et al. Biological markers of sex-based differences in major depressive disorder and in antidepressant response. Eur Neuropsychopharmacol. 2023;76:89-107. doi:10.1016/j.euroneuro.2023.07.012
51. Hosseinzadeh S, Afshari S, Molaei S, Rezaei N, Dadkhah M. The role of genetics and gender specific differences in neurodegenerative disorders: Insights from molecular and immune landscape. J Neuroimmunol. 2023;384:578206. doi:10.1016/j.jneuroim.2023.578206
52. Pujantell M, Altfeld M. Consequences of sex differences in Type I IFN responses for the regulation of antiviral immunity. Front Immunol. 2022;13:986840. doi:10.3389/fimmu.2022.986840
53. Kottilil S, Mathur P. The influence of inflammation on cardiovascular disease in women. Front Glob Womens Health. 2022;3:979708. doi:10.3389/fgwh.2022.979708
54. Sorge RE, LaCroix-Fralish ML, Tuttle AH, et al. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci. 2011;31(43):15450-15454. doi:10.1523/JNEUROSCI.3859-11.2011
55. Elgellaie A, Thomas SJ, Kaelle J, Bartschi J, Larkin T. Pro-inflammatory cytokines IL-1α, IL-6 and TNF-α in major depressive disorder: Sex-specific associations with psychological symptoms. Eur J Neurosci. 2023;57(11):1913-1928. doi:10.1111/ejn.15992
56. Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27(2-3):229-237. doi:10.1016/0165-5728(90)90073-v
57. Swiss Institute of Bioinformatics. Cellosaurus cell line BV-2 (CVCL_0182). Cellosaurus.org. October 5, 2023. Accessed January 26, 2024. https://www.cellosaurus.org/CVCL_0182
58. Jackson SJ, Andrews N, Ball D, et al. Does age matter? The impact of rodent age on study outcomes. Lab Anim. 2017;51(2):160-169. doi:10.1177/0023677216653984
59. Kerkenberg N, Wachsmuth L, Zhang M, et al. Brain microstructural changes in mice persist in adulthood and are modulated by the palmitoyl acyltransferase ZDHHC7. Eur J Neurosci. 2021;54(6):5951-5967. doi:10.1111/ejn.15415
60. Shen Q, Zhang R, Bhat NR. MAP kinase regulation of IP10/CXCL10 chemokine gene expression in microglial cells. Brain Res. 2006;1086(1):9-16. doi:10.1016/j.brainres.2006.02.116